

Firstly, I want to express my sincere congratulations to Zhuo Fan, my graduate mentor, whose paper was recently accepted by 《Angew. Chem. Int. Ed.》! This project has been a fascinating work for all of us involved, including Yuanyuan Xu, Kunxu Teng, Professor Yang, and Professor Niu. Looking back, I see it as an inspiring and exploratory journey that deepened my enthusiasm for this field and allowed me to contribute to the synthesis work and the writing of supporting information. So I wanna write this blog to introduce and look back this work.
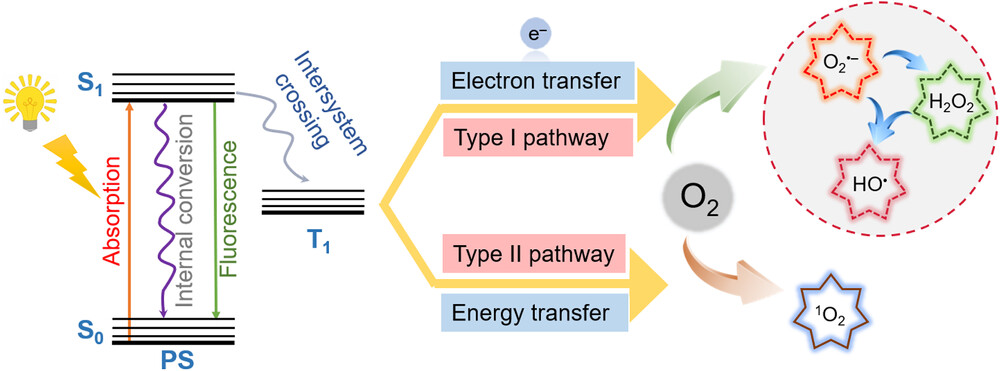
Photodynamic therapy (PDT) is a promising cancer treatment method that employs photosensitizers (PSs), molecular oxygen and light to generate cytotoxic reactive oxygen species (ROS) that kill cancer cells. PDT has been used in the treatment of a variety of tumors (e.g. skin, esophageal and bladder cancers). With advancements in endoscopy and fiber-optic technologies, PDT holds great potential for the treatment of solid tumors, including gastric, liver and cervical cancers. However, hypoxia, a common trait of malignant solid tumors, poses a challenge to PDT efficacy due to its heavy reliance on oxygen concentration. Addressing this challenge, the development of hypoxia-tolerant PSs that maintain high phototoxicity in hypoxic environments is crucial for improving PDT efficacy to solid tumors. Recent advancements have demonstrated that Type-I PDT exhibits reduced dependence on oxygen concentration compared with traditional Type-II PDT. Type-I PSs not only generate ROS (e.g., superoxide radicals and hydroxyl radicals) but also directly damage biomolecules through electron/hydrogen atom transfer between excited PSs and biological substrates.
Designing effective Type I PSs is challenging because various excited-state decay processes compete with energy transfer. One design strategy is to introduce electron donors or acceptors to enhance electron transfer efficiency. However, achieving a balance between efficient electron transfer and maintaining high oxidation potential is complex. Strong electron donors, while enabling effective electron transfer, can lower the PS’s oxidation potential, thus reducing its ability to oxidize biomolecules effectively.
Beginning of this idea
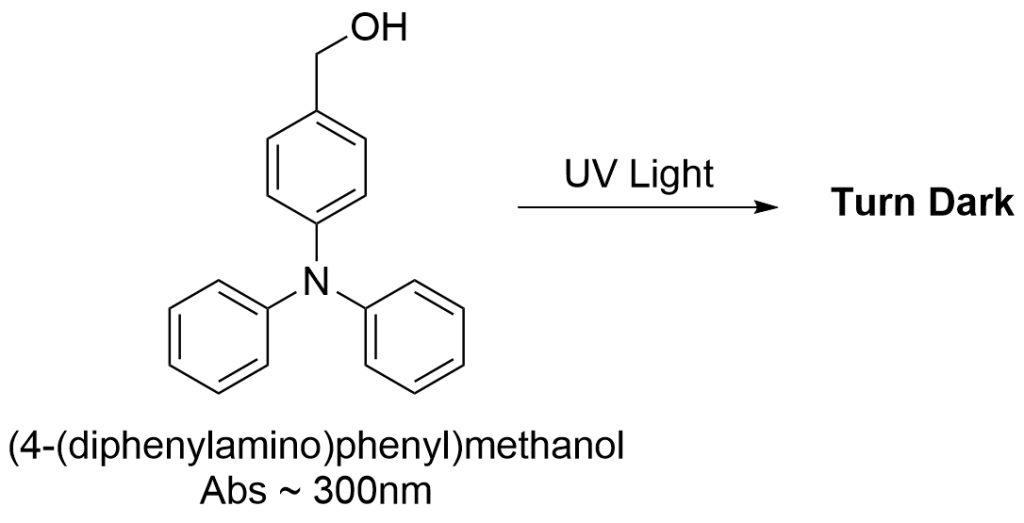
Our idea to address this balance emerged from an unexpected observation: during a synthesis of a BODIPY-triphenylamine derivative, we found that the compound turned dark under UV light on TLC plates, likely due to the reactive benzyl position of triphenylamine. This reminded us of an earlier study where we synthesized a polymer containing both a photosensitizer monomer and an electron donor monomer, which, interestingly, also had a benzyl position. Unexpectedly, this polymer demonstrated exceptional cytotoxicity, but we didn’t figure it out then. Upon realizing that both might be linked to the highly reactive benzyl position, we searched for references and sketched a proposed catalytic cycle that could explain this unusual reactivity. We hypothesized that a covalent bond between PS and triphenylamine could enable a photoinduced electron transfer, generating BODIPY anion and triphenylamine cation radicals. The BODIPY anion radical could transfer electrons to oxygen, creating O₂⁻•, while the benzyl cation radical could oxidize some biomolecules through hydrogen atom transfer.
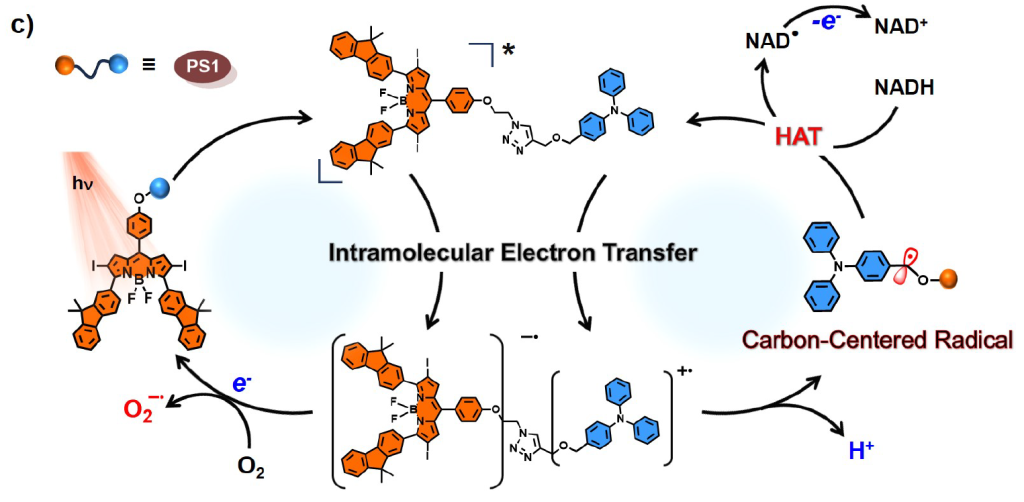
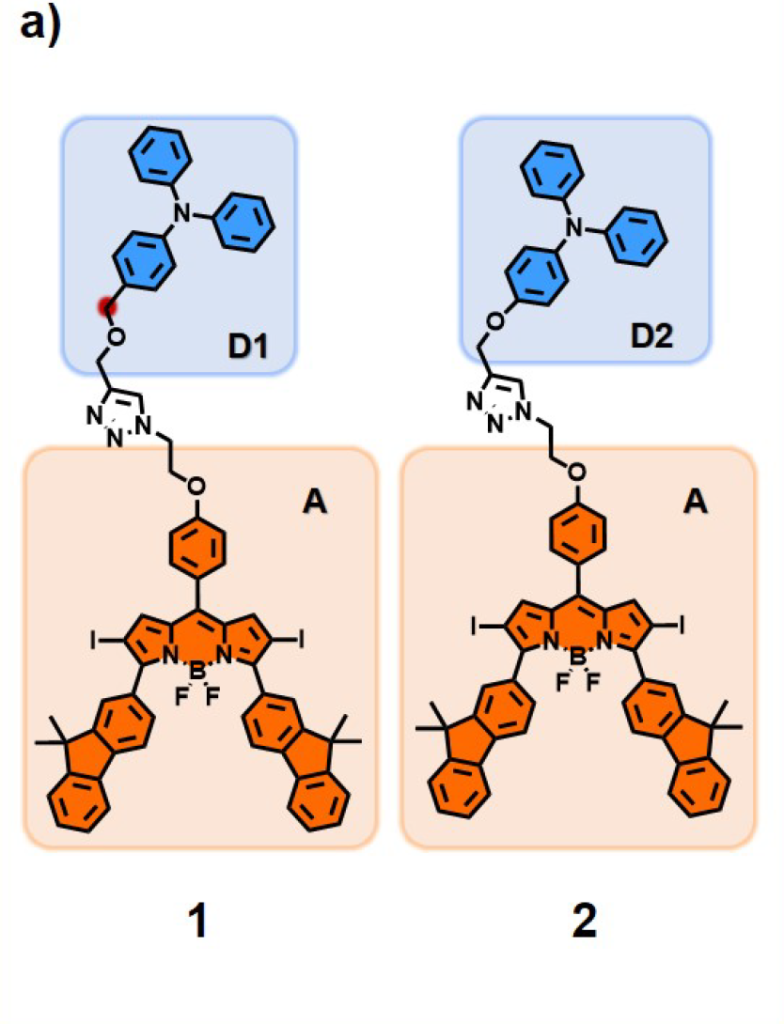
Guided by this idea, we synthesized our target molecule, BFBnT (Bodipy-Fluorene-Bn-TPA). The triphenylamine group acts as the electron donor, while the BODIPY core, modified with fluorene at the 3,5 positions to maximize absorption to PDT wavelength window and iodine at the 2,6 positions for intersystem crossing efficiency(ICT), serves as the electron acceptor. After further optimization of the linker structure through click chemistry, we synthesized a control molecule, BFT (Bodipy-Fluorene-TPA), without the benzyl position.
Synthesis of target molecule
Our synthesis begins with a Williamson ether synthesis reaction, where we reacted p-hydroxybenzaldehyde with an excess of 1-bromo-2-chloroethane to introduce the desired ether linkage. Afterward, we conducted a reaction with pyrrole in the presence of trifluoroacetic acid (TFA) as a acid catalyst, allowing it to proceed overnight to form a dipyrromethane intermediate.
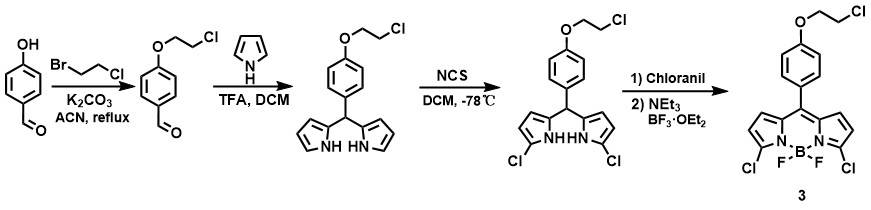
Next, at a low temperature of -78°C, we performed selective chlorination on the pyrrole ring using N-chlorosuccinimide (NCS). This was followed by oxidation with tetrachloro-p-benzoquinone for one hour. Then we added triethylamine and coordinated boron trifluoride etherate, forming the dichloro-BODIPY core structure (compound 3).
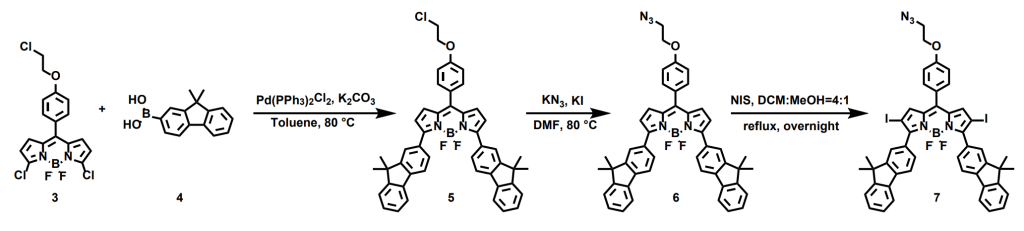
To modify this core, we utilized the Suzuki-Miyaura coupling reaction to introduce fluorene groups at the 3 and 5 positions, enhancing the compound’s conjugation. In the next step, azide groups were introduced through nucleophilic substitution. Lastly, we iodinated the electron-rich 2 and 6 positions using N-iodosuccinimide (NIS) to introduce heavy atom effect, further improving intersystem crossing efficiency, completing the Bodipy synthesis part.
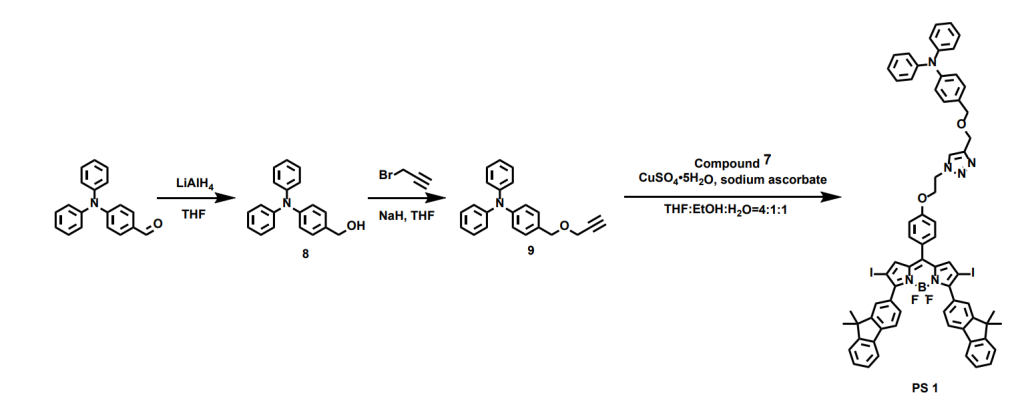
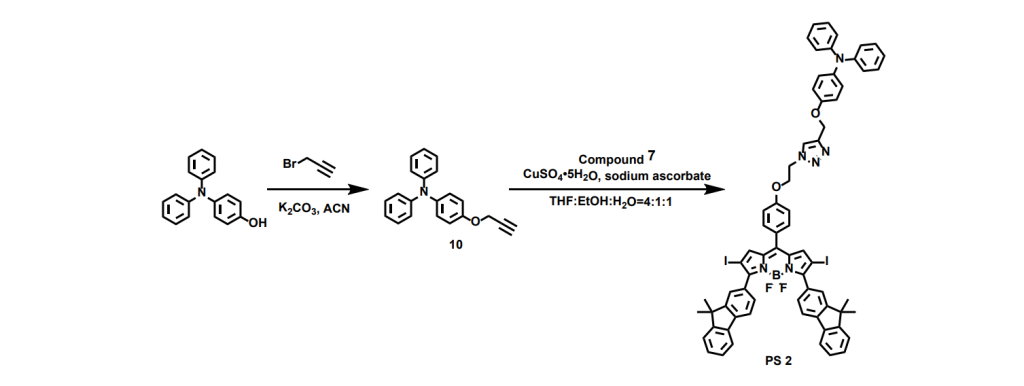
In the synthesis of the triphenylamine segment, we began by introducing an alkyne group through a nucleophilic substitution reaction. This allowed us to connect it with the azide-functionalized BODIPY block via a click reaction—a coupling method I especially enjoy for its efficiency and reliability. Then we get the target molecule BFBnT(PS 1) and the control molecule BFT(PS 2).
Reveal the HAT mechanism behind
Next, we performed detailed mechanistic characterization to understand the reaction pathway. To confirm the formation of carbon-centered radicals, we utilized radical trapping, high-resolution mass spectrometry (HRMS), and electron spin resonance (ESR) spectroscopy.
We explored methods to further confirm the presence of intermediate carbon radicals. For example, we attempted to detect these intermediates using deuterated NADH analogs in a hydrogen-deuterium exchange experiment.
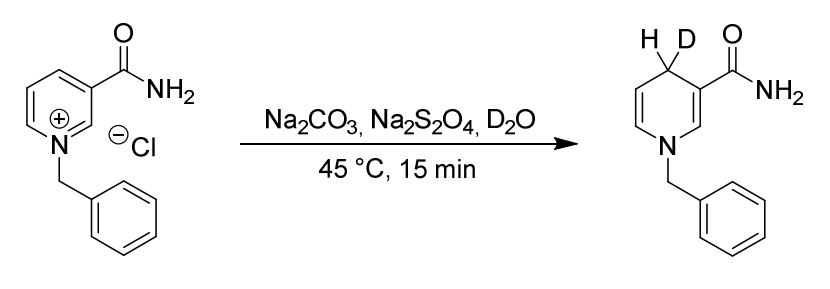
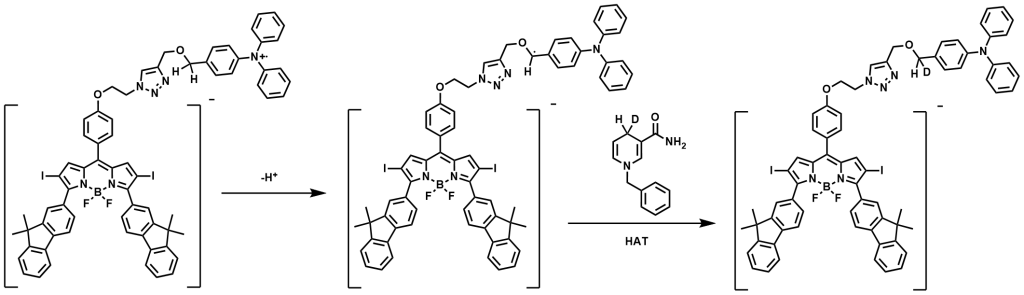
we aimed to capture deuterium-substituted BFBnT at the benzyl position via H-D exchange experiments. We planned to characterize this process using HRMS and 1H NMR, specifically tracking the decrease in benzyl proton integration after irradiation.
However, sadly, the result was not obvious, so we turned to use spin-trapping techniques.
Using spin-trapping techniques, we employed TEMPO to capture carbon-centered radicals and DMPO to target both carbon and superoxide anion radicals. This approach allowed us to verify the formation of stable radical adducts, which we subsequently analyzed with HRMS and ESR spectroscopy to ensure precise identification.
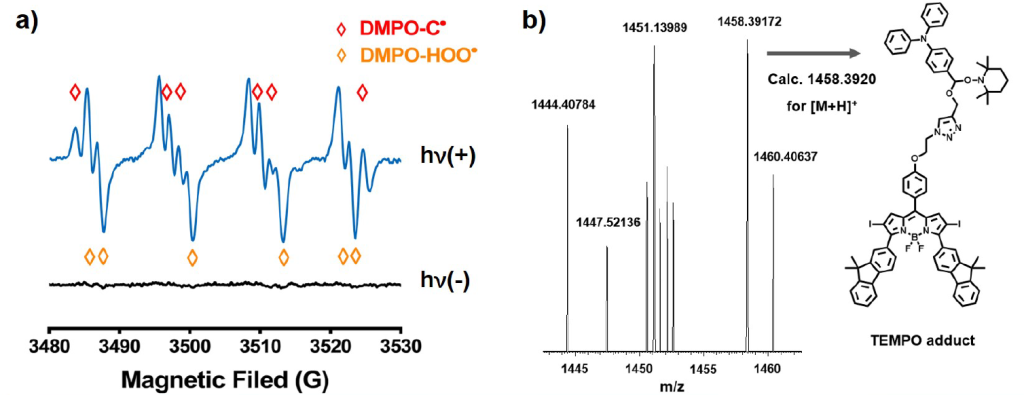
b) HRMS spectrum of the irradiated mixture of PS 1 and TEMPO.
The study of photo-induced electron transfer
Subsequently, we delved into computational and electrochemical analyses to better understand the electron transfer mechanisms.
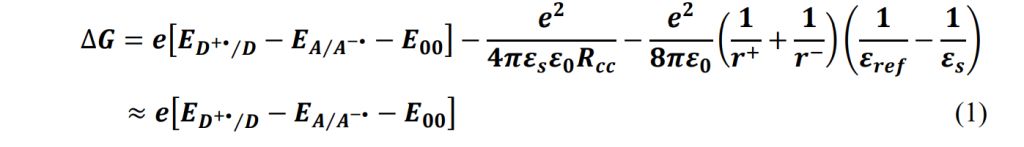
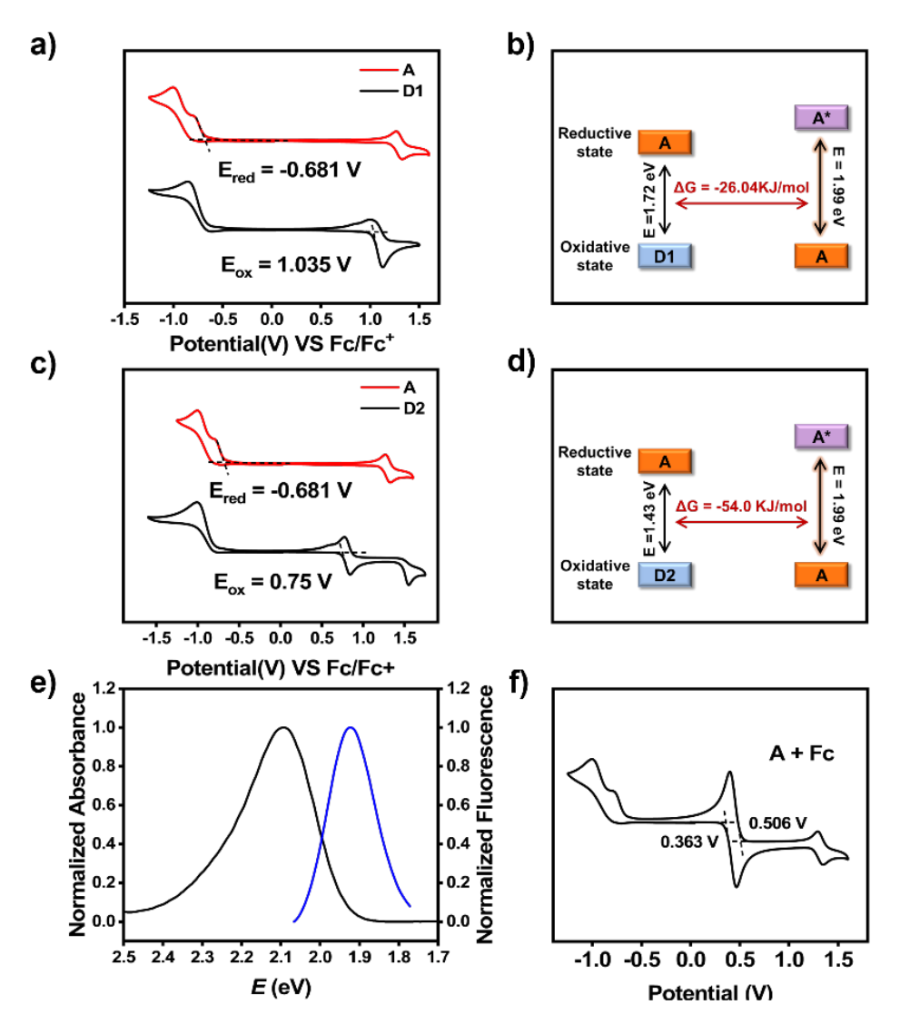
The Gibbs free energy change for electron transfer from donors D1/D2 to acceptor A can be calculated using the Weller equation (1). In this equation, 𝐸𝐷+•⁄𝐷 represents the initial potential for one-electron oxidation of the electron donor, while 𝐸𝐴/𝐴−• is the initial potential for oneelectron reduction of the electron acceptor. By converting the wavelength axis to an energy scale, 𝐸00 can be estimated at the intersection of the normalized absorbance and emission spectra of A.
We then synthesized model compounds A, D1, and D2, followed by detailed electrochemical studies.、
Electrochemical measurements show that D1 has oxidation potentials of +1.035 V vs. Fc/Fc+ , D2 has oxidation potentials of +0.75 V vs. Fc/Fc+ and the A has reduction potentials of -0.681 V vs. Fc/Fc+. The excited state energy E00 is estimated to be 1.99 eV. Rehm-Weller theory calculations reveal that the Gibbs free energy changes (ΔG) for photoinduced electron transfer are thermodynamically favorable: -26.04 kJ/mol for the transfer from excited-state D1 to A , and -54.0 kJ/mol for the transfer from excited-state D2 to A. These results indicate that electron transfer in both processes is feasible.
PDT Application
We encapsulated BFBnT (PS 2) as nanoparticles using Tween 60, followed by a comprehensive evaluation of their reactive oxygen species (ROS) generation capacity, NADH oxidation capabilities, and in vitro and in vivo performance.
1O2 Production – ABDA Assay
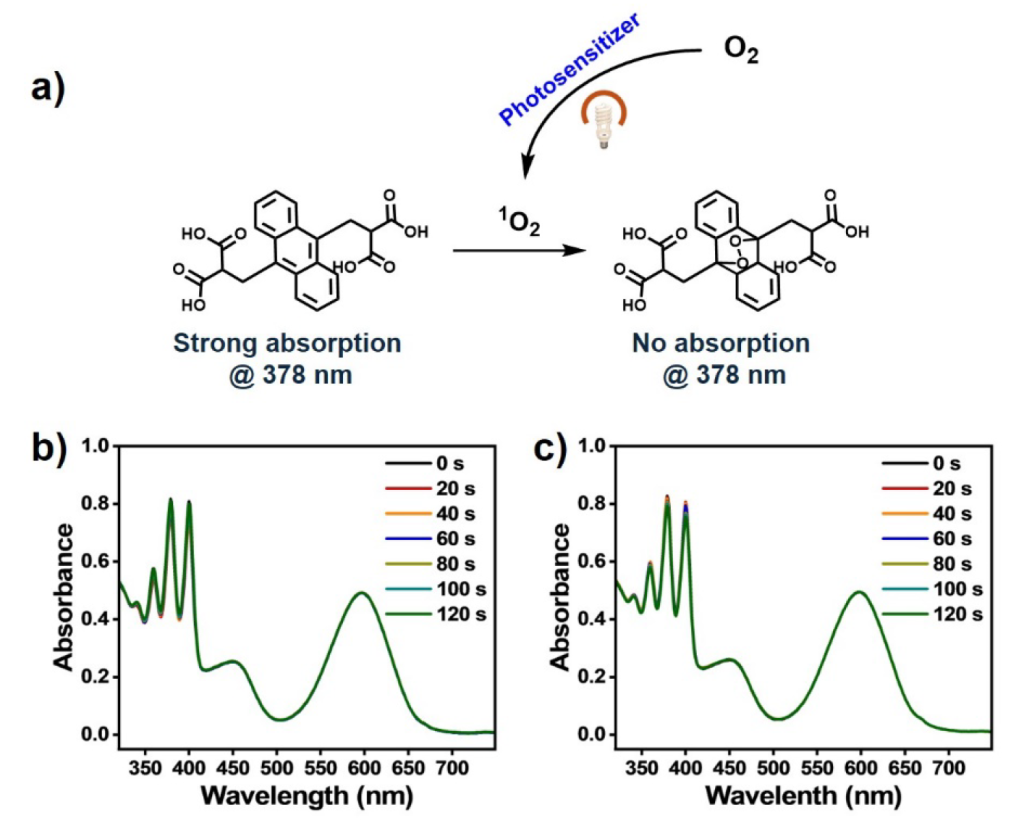
To evaluate 1O2 production, we employed the commercial probe ABDA (anthracene-9,10-dipropionic acid disodium salt), a water-soluble anthracene derivative that reacts with 1O2, resulting in photobleaching to form endoperoxides. This reaction was monitored by observing a characteristic decrease in absorbance around 400 nm in the UV-Vis spectrum.
The results showed minimal 1O2 production, suggesting that the photosensitizer’s efficiency in generating 1O2 through energy transfer is low.
·OH Production – 1,4-Dicarboxybenzene Assay
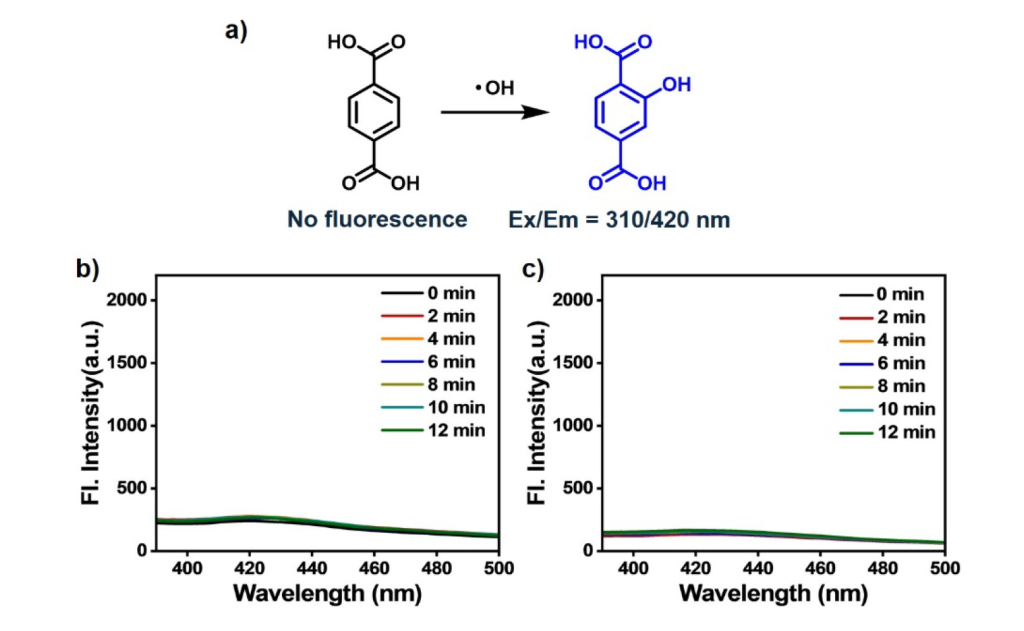
We used 1,4-dicarboxybenzene as a probe for detecting ·OH, which can add to aromatic rings. The formation of ·OH was assessed by measuring fluorescence emissions between 390 nm and 500 nm.
This test indicated negligible ·OH production, confirming that the photosensitizer does not rely on ·OH to oxidize NADH.
O2– Production- DHE Assay
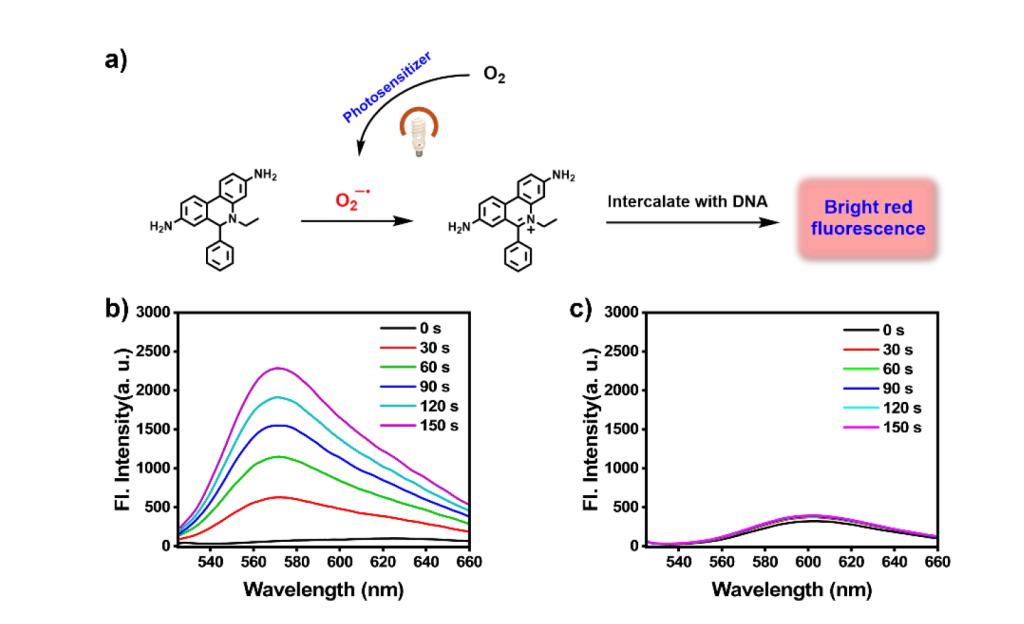
The capability of the photosensitizer to generate O2– was tested using dihydroethidium (DHE), a widely used probe for O2– detection in live cells. Upon cell uptake, DHE is dehydrogenated by intracellular O2– to produce ethidium, which binds to DNA and emits red fluorescence.
Results revealed that BFBnT nanoparticles are highly effective in generating O2–, validating that the photosensitizer primarily operates through an electron transfer pathway under light exposure, which means this is a typical type I PS.
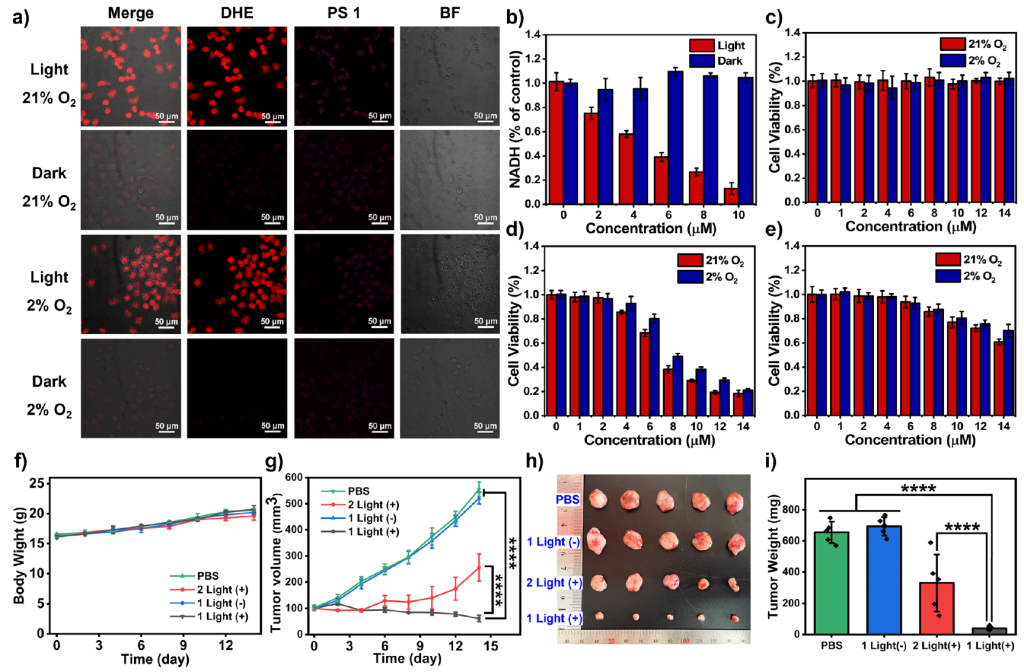
We also did the test of NADH oxidation capabilities, and in vitro and in vivo performance to characterize its photodynamic therapy efficiency.
Conclusion
In summary, this work presents a novel design strategy for Type-I photosensitizer with the involvement of hydrogen atom transfer (HAT). HAT-involved Type-I photosensitizer PS 1 was developed, as a proof of concept. It simultaneously produces carbon-centered radicals to oxidize NADH and generates superoxide radicals under light-irradiation.
Looking back on this truly inspiring project, I’m still filled with a profound sense of excitement and pride. I want to offer my heartfelt congratulations and thanks to Zhuo Fan. He is an exceptionally hardworking chemist, graduate mentor, friend, and teacher. Seeing this work published in Angewandte Chemie is a well-deserved recognition of his dedication and expertise. He derserved it.
This project marked my first complete research experience, and I was able to witness its evolution over the past year—from its initial concept through complex challenges to the final paper. Along the way, I gained deep insights into fields like photocatalysis, electrocatalysis, supramolecular chemistry and photo chemistry. Supporting the synthesis of our target molecule also significantly advanced my organic synthesis skills, and I found great fulfillment in the cycle of identifying, exploring, and solving problems. This journey has not only been enlightening but has also inspired my current research project, guiding my future steps forward.